Deeper look into SampleQC
There are various other tabs in SampleQC, that are relevant for BulkRNA data.
Read distribution
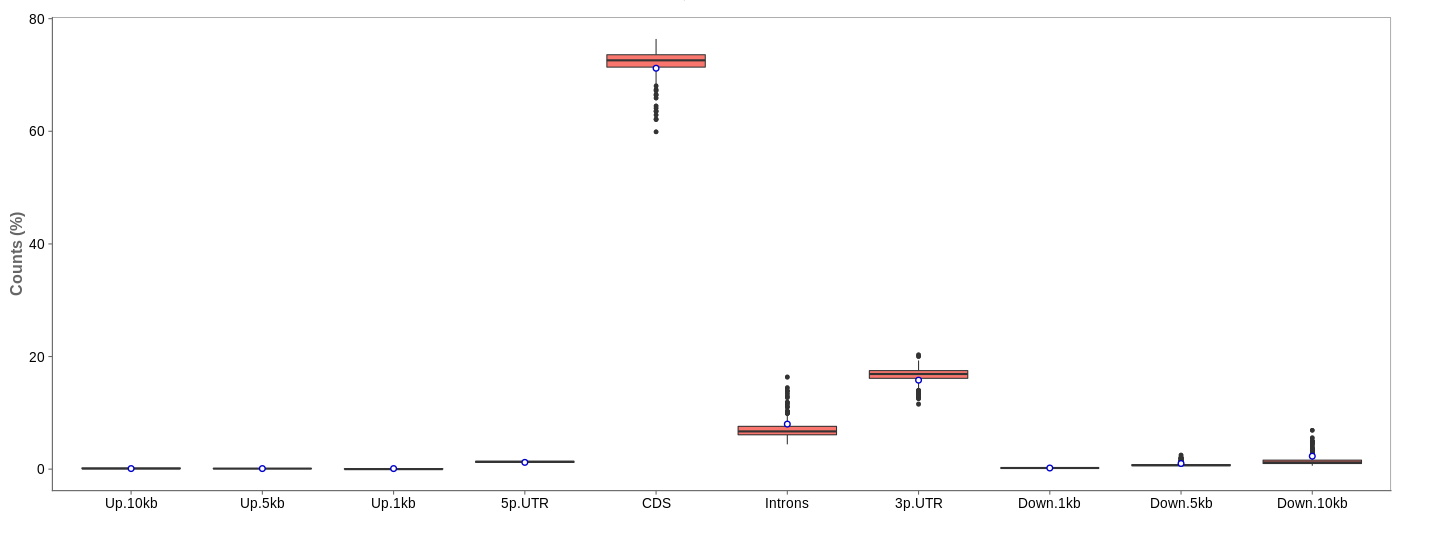
First of all, the box for total percentages should be ticked in the bottom for better comparison.
Ideally, the percentage of counts mapping to the highest should be CDS which stands for coding DNA sequence, relating to the region that codes for protein.
Also important is that up- and downstream are not mapped often.
Gene body coverage
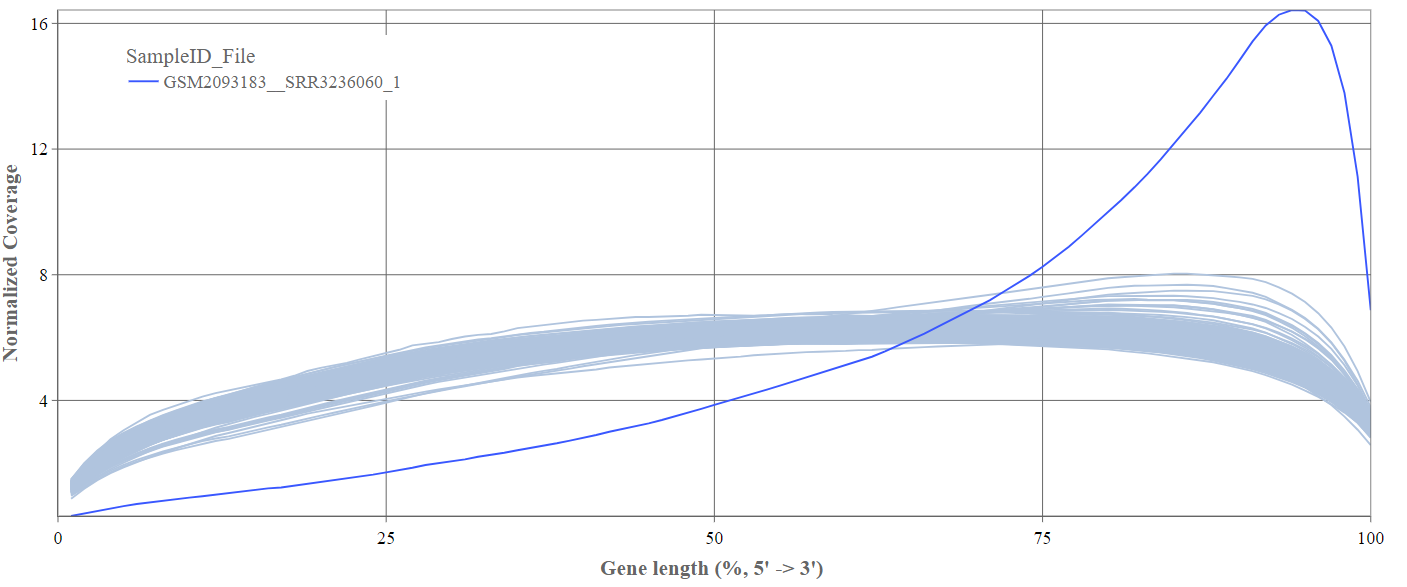
We can see the normalized coverage. The samples gene body coverages should be close to one another and should form a homogeneous band and, in BulkRNA-Seq, should be spanning quite ‘uniform’ over the gene length going down at the start and endpoints. Please note that evoenthouth this QC section is called gene body coverage we effectively measure the coverage along the RNA transcripts corresponding to the gene.
The sample colored in blue shows very untypical behaviour and is advised to be excluded.
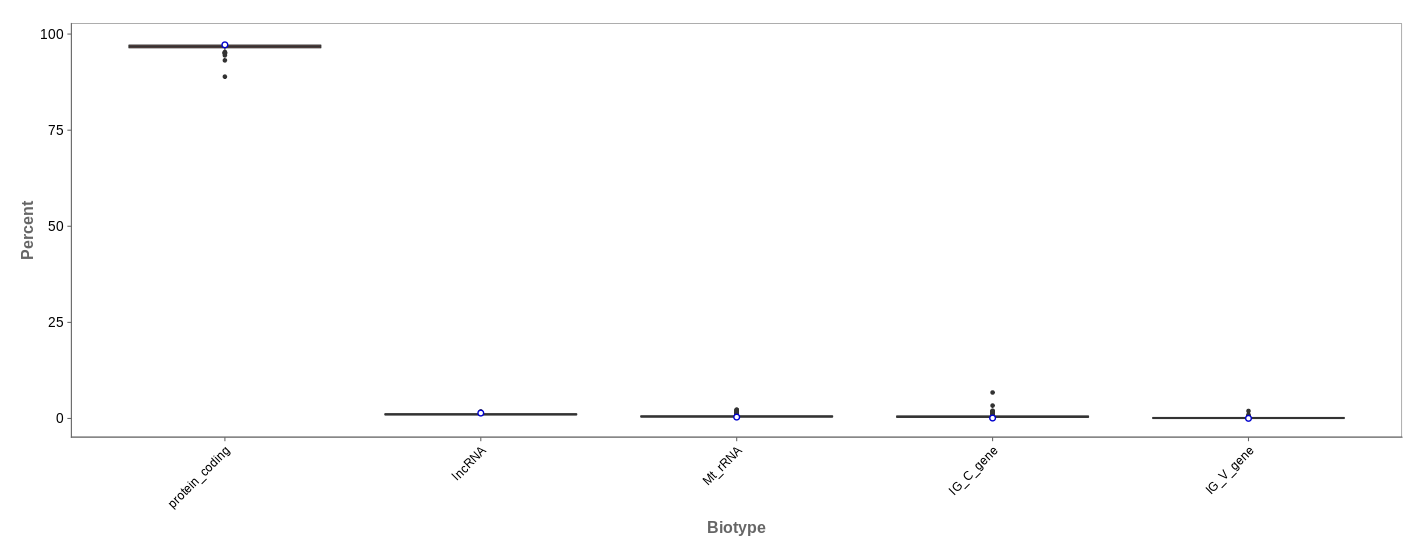
Biotype
The biotype (that is functional types of the genes to which reads are mapped) can be used for quality control. The protein_coding should be the highest and not far away from 100%. We can also see in our example that there we have IgC and IGv genes for tuberculosis data, which seems reasonable because they are relevant for the immune/antibody system of humans.
Mitochondrial
Usually, the proportion of transcripts mapping to mitochondrial genes should be low. If there is phenomenons such as cell death, the amount of mitochondrial transcripts increases and we might infer a lower quality for the data.
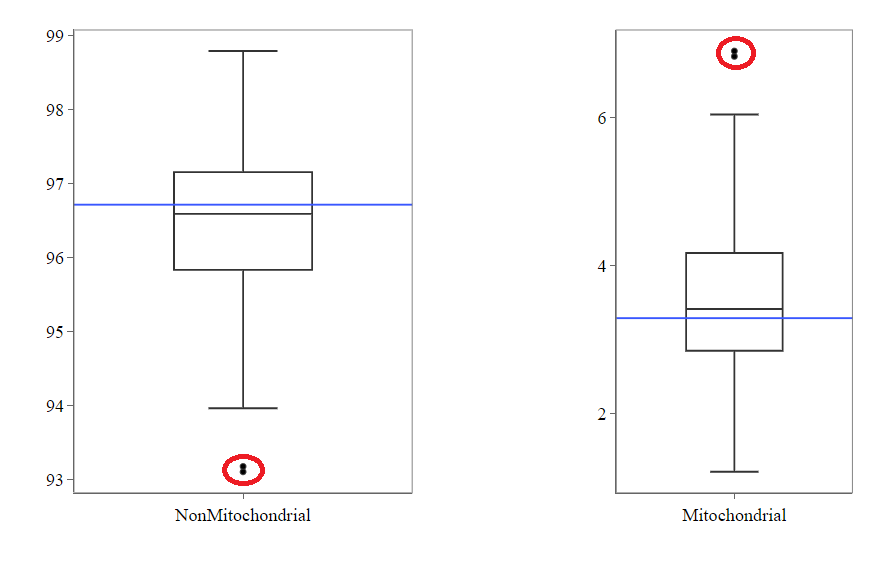
In our example, it may be useful to take out the two circled samples because they are clear outliers from the rest of the data.
Genes
An additional evaluation can be done by looking into the Genes tab, checking which genes are counted the most. The interpretation of this analysis is experiment specific and requires knowledge of the biological system that is investigated.