This the multi-page printable view of this section. Click here to print.
Data Quality Control
- 1: QC parameters for transcriptomics
- 2: Quality Control for Bulk RNA-Seq data
- 2.1: Starting quality control
- 2.2: Deeper look into SampleQC
- 2.3: QC clustering
- 2.4: Finalizing the Quality Control
1 - QC parameters for transcriptomics
Quality control (QC) parameters are crucial for ensuring the accuracy and reliability of RNA sequencing (RNA-Seq) data. This documentation provides an overview of key QC parameters, their importance, and how to interpret them to maintain high data quality throughout RNA-Seq experiments. Note that the interpretation of these parameters can vary based on the experiment conducted and the guidance provided here is a more general guidance.
Alignments - Star
Number of input reads
Description
The number of input reads refers to the total count of raw sequencing reads generated from an RNA-Seq experiment before any filtering or quality control steps are applied. This parameter is crucial as it provides an initial measure of the sequencing depth, which directly impacts the sensitivity and accuracy of downstream analyses. A higher number of input reads generally indicates a more comprehensive representation of the transcriptome, allowing for better detection of low-abundance transcripts and more accurate quantification of gene expression levels.
Interpretation
From a quality control perspective, the number of input reads is essential for assessing the overall success of the sequencing run. If the number of input reads is significantly lower than expected, it may indicate issues such as poor RNA quality, inefficient library preparation, or technical problems during sequencing. Conversely, an excessively high number of input reads might suggest over-sequencing, which could lead to unnecessary costs without substantial gains in data quality.
% uniquely mapped reads
Description
The % uniquely mapped reads refers to the percentage of sequencing reads that align to a single, unique location in the reference genome or transcriptome. This parameter is critical for assessing the specificity and accuracy of the mapping process. High percentages of uniquely mapped reads indicate that the reads are aligning well to the reference, suggesting good quality of the sequencing data and the reference genome. This is important for downstream analyses, such as differential gene expression and variant calling, as it ensures that the data accurately represents the biological sample.
Interpretation
From a quality control perspective, the % uniquely mapped reads can reveal potential issues with the RNA-Seq experiment. Low percentages of uniquely mapped reads may indicate problems such as sample degradation or contamination, poor quality of the reference genome, or the presence of repetitive sequences that cause reads to map to multiple locations. Additionally, it can highlight technical issues like sequencing errors or biases introduced during library preparation. Monitoring this parameter helps in identifying and addressing these issues, ensuring the reliability and accuracy of the RNA-Seq data for further analysis.
% reads mapped to multiple location
Description
The % reads mapped to multiple loci refers to the percentage of sequencing reads that align to more than one location in the reference genome or transcriptome. This parameter is important because it provides insight into the complexity and redundancy of the sequencing data. Reads that map to multiple loci often arise from repetitive sequences, paralogous genes, or regions with high sequence similarity. While some level of multi-mapping is expected, especially in complex genomes, a high percentage of reads mapped to multiple loci can complicate downstream analyses and reduce the accuracy of gene expression quantification.
Interpretation
From a quality control perspective, the % reads mapped to multiple loci can indicate potential issues with the RNA-Seq experiment or the reference genome. High levels of multi-mapping reads may suggest problems such as contamination, sequencing errors, or the presence of highly repetitive sequences that are difficult to resolve. Additionally, it can reflect the quality of the reference genome, as incomplete or poorly annotated references may lead to ambiguous mappings. Monitoring this parameter helps identify and address these issues, ensuring the reliability and interpretability of the RNA-Seq data for further analysis.
Average input read length
Description
The Average input read length refers to the mean length of the sequencing reads generated from an RNA-Seq experiment before any trimming or filtering steps are applied. Longer read lengths generally allow for better alignment to the reference genome or transcriptome, improving the accuracy of downstream analyses such as transcript assembly and gene expression quantification. Additionally, longer reads can span more complex regions of the genome, providing more comprehensive coverage and reducing the likelihood of ambiguous mappings.
Interpretation
The input read length is typically predefined by the sequencing protocol and can be useful for QC purposes when considered in combination with other quality metrics (e.g. Average mapped length)
Average mapped length
Description
The Average mapped length refers to the mean length of the sequencing reads that have been successfully aligned to the reference genome or transcriptome. This parameter is important because it provides insight into the quality of the mapping process and the integrity of the RNA-Seq data. Longer average mapped lengths generally indicate that the reads are aligning well and covering more of the reference sequence, which is beneficial for accurate transcript assembly and gene expression quantification. It also suggests that the sequencing reads are of high quality and have not been excessively fragmented or degraded.
Interpretation
From a quality control perspective, the average mapped length can help identify potential issues with the RNA-Seq experiment. Shorter mapped lengths may indicate problems such as RNA degradation, poor library preparation, or sequencing errors that result in truncated reads. Additionally, discrepancies between the average input read length and the average mapped length can highlight issues with the mapping process, such as the presence of adapter sequences or low-complexity regions that are not being properly aligned.
Number of splice sites total
Description
The Number of splice sites total refers to the total count of unique splice junctions identified in RNA-Seq data. Splice sites are the locations where introns are removed, and exons are joined together during the splicing process. This parameter is crucial for understanding the complexity of the transcriptome, as it provides insights into the diversity of splicing events occurring within a sample. High numbers of splice sites can indicate a rich variety of alternative splicing events, which contribute to the generation of multiple transcript isoforms from a single gene, thereby increasing proteomic diversity.
Interpretation
From a quality control (QC) perspective, the Number of splice sites total can be used to assess the completeness and accuracy of the splicing process captured in the RNA-Seq data. A higher-than-expected number of splice sites might suggest potential issues such as sequencing errors, misalignment, or the presence of novel splicing events that were not previously annotated. Conversely, a lower-than-expected number of splice sites could indicate incomplete splicing, poor RNA quality, or insufficient sequencing depth.
Number of splice sites annotated
Description
The Number of splice sites annotated refers to the total count of splice junctions that align with known, annotated splice sites in a reference genome. These annotated splice sites are critical for understanding the splicing landscape of a sample, as they represent well-characterized regions where introns are excised, and exons are joined during mRNA processing. This parameter is essential for validating the accuracy of RNA-Seq data, as it helps confirm that the observed splicing events correspond to biologically relevant and previously identified splice sites.
Interpretation
From a quality control (QC) perspective, the Number of splice sites annotated can be used to assess the fidelity of the splicing process and the quality of the RNA-Seq data. A high number of annotated splice sites indicates that the sequencing and alignment processes are accurately capturing known splicing events, which is a sign of good data quality. Conversely, a lower-than-expected number of annotated splice sites might suggest issues such as incomplete or degraded RNA, sequencing errors, or alignment problems.
Mismatch rate per base
Description
The Mismatch rate per base refers to the proportion of bases in the RNA-Seq reads that do not match the reference genome at the corresponding positions. This parameter is calculated by dividing the number of mismatched bases by the total number of aligned bases. It is a critical metric for assessing the accuracy of the sequencing and alignment processes. A low mismatch rate indicates high-quality sequencing data and accurate alignment, whereas a high mismatch rate may suggest issues such as sequencing errors, poor-quality reads, or problems with the reference genome.
Interpretation
From a quality control (QC) perspective, the Mismatch rate per base is essential for evaluating the overall reliability of RNA-Seq data. A higher-than-expected mismatch rate can indicate potential problems such as contamination, sequencing artifacts, or alignment errors. It may also reflect biological variations, such as the presence of single nucleotide polymorphisms (SNPs) or RNA editing events. Conversely, an unusually low mismatch rate may result from overly strict alignment settings that discard divergent reads, potentially leading to bias or loss of information.
Deletion rate per base
Description
The Deletion rate per base refers to the proportion of bases in RNA-Seq reads that are deleted relative to the reference genome. This parameter is calculated by dividing the number of deleted bases by the total number of aligned bases. It is an important metric for assessing the quality of the sequencing and alignment processes. A low deletion rate indicates high-quality sequencing data and accurate alignment, whereas a high deletion rate may suggest issues such as sequencing errors, poor-quality reads, or problems with the reference genome.
Interpretation
From a quality control (QC) perspective, the Deletion rate per base is essential for evaluating the overall reliability of RNA-Seq data. A higher-than-expected deletion rate can indicate potential problems such as contamination, sequencing artifacts, or alignment errors. It may also reflect biological variations, such as the presence of indels (insertions and deletions) in the sample. Conversely, an unusually low deletion rate may result from overly strict alignment settings that discard divergent reads, potentially leading to bias or loss of information.
Insertion rate per base
Description
The Insertion rate per base refers to the proportion of bases in RNA-Seq reads that are inserted relative to the reference genome. This parameter is calculated by dividing the number of inserted bases by the total number of aligned bases. It is an important metric for assessing the quality of the sequencing and alignment processes. A low insertion rate indicates high-quality sequencing data and accurate alignment, whereas a high insertion rate may suggest issues such as sequencing errors, poor-quality reads, or problems with the reference genome.
Interpretation
From a quality control (QC) perspective, the Insertion rate per base is essential for evaluating the overall reliability of RNA-Seq data. A higher-than-expected insertion rate can indicate potential problems such as contamination, sequencing artifacts, or alignment errors. It may also reflect biological variations, such as the presence of indels (insertions and deletions) in the sample. Conversely, an unusually low insertion rate may result from overly strict alignment settings that discard divergent reads, potentially leading to bias or loss of information.
Alignments – Star solo & CBquant file based
Total reads
Description
The Total reads parameter refers to the number of RNA-Seq reads in the processed FASTQ file. This parameter is a fundamental metric for assessing the depth and coverage of the sequencing experiment. It provides an overall measure of the amount of data produced, which is crucial for downstream analyses. A higher number of total reads generally indicates better coverage of the transcriptome, allowing for more accurate and reliable results. Total reads is the sum of Matching reads, Hamming reads and Not Reads.
Interpretation
From a quality control (QC) perspective, the total reads parameter is essential for evaluating the efficiency and success of the RNA-Seq experiment. A sufficient number of reads is necessary to ensure that all expressed genes are adequately represented in the data. If the total reads count is lower than expected, it may indicate issues such as poor RNA quality, inefficient library preparation, or sequencing issues. Conversely, an excessively high number of reads can lead to unnecessary costs without significant gains in data quality. Therefore, interpreting the total reads parameter in the context of the experimental design and the specific goals of the study is crucial for optimizing the balance between data quality and resource utilization.
Matching reads
Description
The Matching reads parameter refers to the number of RNA-Seq reads whose barcode sequences exactly match one of the reference barcodes. Barcodes are short, unique sequences added to each RNA molecule during library preparation to identify and differentiate between samples in multiplexed sequencing experiments. This parameter is crucial for ensuring the accuracy of sample identification and the integrity of the sequencing data, as it confirms that the reads have been correctly assigned to their respective samples.
Interpretation
From a quality control (QC) perspective, the Matching reads parameter is essential for evaluating the efficiency and accuracy of the barcode assignment process. A high number of matching reads indicates that the barcoding and sequencing processes were successful, with minimal errors in sample identification. Conversely, a lower number of matching reads might suggest issues such as barcode misassignment, sequencing errors, or contamination between samples. By analysing the proportion of matching reads, researchers can identify potential problems and ensure the reliability of the ScreenSeq data.
Hamming reads
Description
The Hamming reads parameter refers to the number of RNA-Seq reads that match the reference sequence with a single base mismatch, i.e., Hamming distance of 1. The Hamming distance is a measure of sequence similarity, defined as the number of differing bases between two sequences of equal length. In the context of RNA-Seq, this parameter helps in assessing the accuracy of read alignment by quantifying the number of reads that closely match the reference genome, allowing for one base mismatch.
Interpretation
From a quality control (QC) perspective, the Hamming reads parameter is crucial for evaluating the fidelity of the sequencing and alignment processes. A higher number of Hamming reads indicates a greater frequency of errors in the barcode sequences and suggests lower data quality.
Not Reads
Description
The Not Reads parameter refers to the number of remaining RNA-Seq reads that could not be matched to any expected barcode. These unmatched reads are typically excluded from downstream analyses as they cannot be attributed to any sample.
Interpretation
The increased fraction of NotReads can result from various factors such as sequencing or library preparation issues and is a potential indicator of lower data quality.
Percent Demux
Description
The Percent Demux parameter refers to the percentage of RNA-Seq reads that have been successfully demultiplexed, calculated as the sum of “Matching Reads” and “Hamming Reads” divided by “Total Reads”. Demultiplexing is the process of assigning reads to their respective samples based on unique barcode sequences. This parameter is crucial for assessing the efficiency and accuracy of the demultiplexing process, ensuring that reads are correctly attributed to their corresponding samples.
Interpretation
From a quality control (QC) perspective, the Percent Demux parameter is essential for evaluating the overall success of the sequencing experiment. A high Percent Demux value indicates that the majority of reads have been accurately assigned to their respective samples. Conversely, a lower Percent Demux value might indicate issues such as sequencing errors.
Alignments – Star solo & CBquant demultiplexed
Reads
Description
The Reads parameter in RNA-Seq data refers to the sum of demultiplexed reads, including both uniquely aligned reads and those that could not be uniquely aligned to exon features. This parameter represents the raw sequencing output, encompassing all sequences obtained from the RNA molecules in the sample. Demultiplexing attributes these reads based on their barcodes, which are unique identifiers assigned to each sample during library preparation, ensuring accurate attribution to their respective samples.
Interpretation
From a quality control (QC) perspective, the Reads parameter is fundamental for evaluating the initial quality of RNA-Seq library. In most of the cases a relatively even distribution of reads across samples within one library can be expected. Samples with unexpectedly high or low read counts may indicate certain technical or biological differences between samples (such as issues with library preparation, low number of cells or toxic treatment).
Counts
Description
The Counts parameter in RNA-Seq data refers to the final sum of deduplicated Unique Molecular Identifier (UMI) counts per sample or barcode. This parameter is crucial as it represents the number of unique RNA molecules captured and sequenced in a given sample, providing a quantitative measure of gene expression. Deduplication using UMIs helps to eliminate PCR duplicates, ensuring that each count corresponds to an original RNA molecule, thus enhancing the accuracy of the data.
Interpretation
From a quality control (QC) perspective, the Counts parameter is essential for evaluating the true abundance of transcripts in the sample. In most of the cases a relatively even distribution of UMI counts across samples within one library can be expected. Samples with unexpectedly high or low UMI counts may indicate certain technical or biological differences between samples (such as issues with library preparation, low number of cells or toxic treatment).
ExactM
Description
The ExactM parameter in RNA-Seq data refers to the percentage of exactly matching barcode sequences compared to the sum of all matched barcodes, which includes exact matches and one base mismatches. This parameter is crucial for assessing the accuracy of barcode assignment during the demultiplexing process.
Interpretation
Low ExactM percentages indicates low barcode sequence fidelity and suggest potential issues with sequencing or library preparation.
Unmapped reads, Multimapped reads & Uniquely mapped reads
Description
The Unmapped reads, Multimapped reads, and Uniquely mapped reads parameters in RNA-Seq data refer to the different categories of read alignment outcomes. Unmapped reads are sequences that could not be aligned to the reference genome, often due to sequencing errors, low-quality reads, or the presence of novel sequences not represented in the reference. Multimapped reads are sequences that align to multiple locations in the genome, which can occur in regions with repetitive sequences or homologous genes. Uniquely mapped reads are sequences that align to a single, specific location in the genome, providing precise information about the origin of the RNA molecules. The sum of these three parameters constitutes the total Reads for a sample or barcode.
Interpretation
From a quality control (QC) perspective, the distribution of Unmapped reads, Multimapped reads, and Uniquely mapped reads is crucial for assessing the overall quality and reliability of RNA-Seq data. A high proportion of Uniquely mapped reads indicates accurate sequencing and alignment, essential for reliable transcript quantification and downstream analyses. Conversely, a high proportion of Unmapped reads may suggest issues such as poor RNA quality, sequencing errors, or an incomplete reference genome. A significant number of Multimapped reads can complicate data interpretation, as it may be challenging to determine the exact origin of these reads.
Mapped to Gene, Mapped to no feature, Ambiguously Mapped
Description
The Mapped to Gene, Mapped to no feature and Ambiguously Mapped parameters in RNA-Seq data refer to the different categories of read alignment outcomes within uniquely mapped reads. Mapped to Gene reads are sequences that align to annotated gene regions, providing direct information about gene expression levels. Mapped to no feature reads are sequences that align to the genome but do not correspond to any annotated features, which may include intergenic/intronic regions or unannotated transcripts. Ambiguously Mapped reads are sequences that align to multiple overlapping features, making it difficult to determine their exact origin. The sum of these three parameters constitutes the total number of uniquely mapped reads for a sample or barcode.
Interpretation
From a quality control (QC) perspective, the distribution of Mapped to Gene, Mapped to no feature, and Ambiguously Mapped reads is crucial for understanding the overall quality and reliability of RNA-Seq data. A high proportion of Mapped to Gene reads indicates successful alignment to known gene regions, essential for accurate transcript quantification and downstream analyses. Conversely, a high proportion of Mapped to no feature reads may suggest library preparation issues, incomplete genome annotation or the presence of novel transcripts. A significant number of Ambiguously Mapped reads can complicate data interpretation, as it may be challenging to determine the exact origin of these reads.
% Count Reads
Description
The % Count Reads parameter in RNA-Seq data represents the percentage of deduplicated Unique Molecular Identifier (UMI) counts relative to the total number of demultiplexed reads. This metric provides insight into the proportion of reads that contribute to the final count of unique RNA molecules, reflecting the efficiency of the sequencing and deduplication processes.
Interpretation
From a quality control (QC) perspective, the % Count Reads parameter is essential for evaluating the effectiveness of the sequencing and deduplication processes. A higher percentage indicates that a significant portion of the reads are unique and contribute to the final count, suggesting good library complexity and efficient deduplication. Conversely, a lower percentage may indicate issues such as high levels of PCR duplicates, poor RNA quality, or inefficient library preparation. Note that this parameter also depends on the sequencing depth, as deeper sequencing typically leads to higher sequence deduplication levels.
Number of Genes Identified
Description
The Number of Genes Identified parameter refers to the total count of distinct genes detected in a given sample. This metric is derived from the alignment and quantification processes, where reads are mapped to the reference genome and assigned to specific gene annotations. The number of genes identified provides a comprehensive overview of the transcriptomic diversity present in the sample, reflecting the breadth of gene expression captured during sequencing. The gene is considered to be identified if it has at least certain minimum number of UMIs attributed to it (the default value is 3).
Interpretation
From a quality control (QC) perspective, the Number of Genes identified is crucial for assessing the completeness and depth of the RNA-Seq experiment. A high number of identified genes indicates robust sequencing depth and effective library preparation, capturing a wide range of expressed genes, including those with low abundance. Conversely, a low number of identified genes may suggest issues such as poor RNA quality, insufficient sequencing depth, or suboptimal library preparation.
Read distribution
CDS
Description
CDS refer to the proportion of RNA-Seq reads that map to the corresponding genomic features of annotated transcripts. CDS regions are the parts of the genome that are translated into proteins, making them crucial for understanding gene expression and functional genomics. This parameter helps in assessing how well the RNA-Seq reads cover the protein-coding regions, which is important for accurate quantification of gene expression levels.
Interpretation
From a quality control (QC) perspective, the distribution of reads in CDS is essential for evaluating the efficiency and accuracy of the RNA-Seq experiment. A high proportion of reads mapping to the CDS regions indicates that the sequencing data is effectively capturing the protein-coding transcripts, which is a sign of good data quality. Conversely, a lower-than-expected proportion of reads in the CDS regions might suggest issues such as poor RNA quality, inefficient library preparation, or biases in the sequencing process.
Introns
Description
Introns refers to the proportion of RNA-Seq reads that map to intronic regions of the genome. Introns are non-coding sequences that are removed during the RNA splicing process, and their presence in RNA-Seq data can provide insights into various aspects of gene expression and regulation. This parameter is important for understanding the extent of intron retention, which can occur due to alternative splicing events or incomplete splicing. Analysing the distribution of reads in intronic regions helps in identifying potential regulatory mechanisms and understanding the complexity of the transcriptome.
Interpretation
From a quality control (QC) perspective, Introns is essential for evaluating the efficiency of the splicing process and the quality of the RNA-Seq data. A higher-than-expected proportion of reads mapping to intronic regions might indicate issues such as incomplete splicing, poor RNA quality, or sequencing artifacts. It may also reflect biological phenomena such as intron retention, which can be associated with specific cellular conditions or diseases. Conversely, a lower-than-expected proportion of reads in intronic regions might suggest efficient splicing and high-quality RNA.
3`UTR
Description
3' UTR refers to the proportion of RNA-Seq reads that map to the 3’ untranslated regions (3’ UTRs) of the genome. The 3’ UTR is a segment of mRNA that follows the coding sequence and precedes the poly(A) tail. It plays a crucial role in post-transcriptional regulation of gene expression, including mRNA stability, localization, and translation efficiency. Analysing the distribution of reads in the 3’ UTRs helps in understanding the regulatory mechanisms that control gene expression and the potential impact of alternative polyadenylation events.
Interpretation
From a quality control (QC) perspective, the distribution of reads in the 3' UTR is essential for evaluating the completeness and accuracy of the RNA-Seq data. A higher-than-expected proportion of reads mapping to the 3’ UTRs might indicate the presence of alternative polyadenylation sites, which can lead to the production of mRNA isoforms with different regulatory properties. Conversely, a lower-than-expected proportion of reads in the 3’ UTRs could suggest issues such as incomplete library preparation, degradation of RNA, or biases in the sequencing process. For ScreenSeq, reads mapped to 3’ UTRs represent the largest fraction of all reads mapped to genes.
5’ UTR
Description
5' UTR refers to the proportion of RNA-Seq reads that map to the 5’ untranslated regions (5’ UTRs) of the genome. The 5’ UTR is a segment of mRNA located upstream of the coding sequence and plays a crucial role in the regulation of gene expression. It influences mRNA stability, translation efficiency, and subcellular localization. Analysing the distribution of reads in the 5’ UTRs helps in understanding the regulatory mechanisms that control gene expression, and the potential impact of alternative transcription start sites and upstream open reading frames (uORFs).
Interpretation
From a quality control (QC) perspective, the distribution of reads in the 5' UTR is essential for evaluating the completeness and accuracy of the RNA-Seq data. A higher-than-expected proportion of reads mapping to the 5’ UTRs might indicate the presence of alternative transcription start sites or regulatory elements that affect mRNA stability and translation. Conversely, a lower-than-expected proportion of reads in the 5’ UTRs could suggest issues such as incomplete library preparation, degradation of RNA, or biases in the sequencing process.
Up 10kb, Up 5 kb, Up 1 kb, Down 1kb, Down 5kb & Down 10kb
Description
The Up 10kb, Up 5kb & Up 1kb parameters refer to the proportion of RNA-Seq reads that map to regions upstream of transcription start sites (TSS) at distances of 10kb, 5kb, and 1kb, respectively. These upstream regions are crucial for understanding the regulatory elements that control gene expression, such as promoters, enhancers, and other transcription factor binding sites.
In contrast, the Down 1kb, Down 5kb & Down 10kb parameters refer to the proportion of RNA-Seq reads that map to regions downstream of transcription end sites (TES) at distances of 1kb, 5kb, and 10kb, respectively. These downstream regions are important for understanding the regulatory elements that influence gene expression, such as terminators and other transcription factor binding sites.
Analysing the distribution of reads in these regions helps in identifying potential regulatory mechanisms and understanding how gene expression is modulated in different conditions.
Interpretation
From a quality control (QC) perspective, the distribution of upstream and downstream reads is essential for evaluating the completeness and accuracy of the RNA-Seq data. A higher-than-expected proportion of reads mapping to upstream & downstream regions might indicate the presence of active regulatory elements, which can influence gene expression. Conversely, a lower-than-expected proportion of reads in these regions could suggest issues such as incomplete library preparation, RNA degradation, or biases in the sequencing process. Therefore, interpreting these parameters in the context of the experimental design and known biological expectations is crucial for ensuring the reliability and validity of RNA-Seq data analysis. Additionally, considering these upstream and downstream regions together provides a comprehensive view of the regulatory landscape, helping to identify potential interactions between different regulatory elements and their impact on gene expression.
Mitochondrial
Non MT genes
Description
The non MT genes genes parameter refers to the proportion of RNA-Seq reads that map to nuclear-encoded genes, excluding those encoded by the mitochondrial genome. This parameter is crucial for understanding the expression profile of the nuclear genome, which encompasses the majority of the cellular genes responsible for various biological functions, including metabolism, cell cycle regulation, and signal transduction. By focusing on non-mitochondrial genes, researchers can gain insights into the overall transcriptional activity and identify differentially expressed genes that may be involved in specific cellular processes or disease states.
Interpretation
From a quality control (QC) perspective, the distribution of reads mapping to non-mitochondrial genes is essential for evaluating the completeness and accuracy of the RNA-Seq data. A higher-than-expected proportion of reads mapping to non-mitochondrial genes indicates that the sequencing data effectively captures the nuclear transcriptome, which is a sign of good data quality. Conversely, a lower-than-expected proportion of reads in non-mitochondrial genes might suggest issues such as contamination with mitochondrial RNA, poor RNA quality, or biases in the sequencing process.
MT genes
Description
The MT genes parameter refers to the proportion of RNA-Seq reads that map to genes encoded by the mitochondrial genome. Mitochondrial genes are essential for cellular energy production, as they encode components of the oxidative phosphorylation pathway. These genes are typically highly expressed in cells with high metabolic activity. Monitoring the expression of mitochondrial genes is crucial for understanding cellular metabolism and identifying potential issues related to mitochondrial function.
Interpretation
From a quality control (QC) perspective, the distribution of reads mapping to mitochondrial genes is essential for evaluating the integrity and quality of the RNA-Seq data. A higher-than-expected proportion of reads mapping to mitochondrial genes can indicate poor sample quality, such as a high fraction of apoptotic or lysing cells, which release mitochondrial RNA into the cytoplasm. This can also be observed in samples with increased metabolic activity or necrosis, such as tumor biopsies. Conversely, a lower-than-expected proportion of reads in mitochondrial genes might suggest issues with library preparation or sequencing biases.
Spike in
Description
The SpikeIn parameter refers to the use of synthetic RNA molecules of known sequence and quantity that are added to RNA-Seq samples during library preparation. These spike-in transcripts serve as internal controls to calibrate and normalize the measurements obtained from RNA-Seq experiments. By providing a consistent reference point, spike-ins help in assessing the accuracy and efficiency of the sequencing process, enabling researchers to account for technical variations and biases that may arise during sample preparation, sequencing, and data analysis.
Interpretation
From a quality control (QC) perspective, spike-in transcripts are essential for evaluating the performance of RNA-Seq experiments. They allow for the detection of systematic errors and biases, such as differences in library preparation efficiency or sequencing depth across samples. By comparing the observed expression levels of spike-in transcripts to their known quantities, a researcher can identify and correct for technical artifacts, ensuring that the data accurately reflects the biological variation in the samples. Additionally, spike-ins can be used to assess the sensitivity and dynamic range of RNA-Seq assays, providing valuable information on the reliability and reproducibility of the results.
Biotypes
Biotypes
Description
Biotypes are categories that describe the functional characteristics of genes, such as protein-coding genes, long non-coding RNAs (lncRNAs), microRNAs (miRNAs), small nucleolar RNAs (snoRNAs), ribosomal RNAs (rRNAs) and others. In RNA-Seq data, this refer to the classification of reads based on the biotypes of the genes they map to. These parameters are crucial for understanding the composition and functional diversity of the transcriptome, as they provide insights into the different types of RNA molecules present in a sample and their potential roles in cellular processes.
Interpretation
From a quality control (QC) perspective, analysing the distribution of reads across different biotypes is essential for evaluating the completeness and accuracy of the RNA-Seq data. A balanced distribution of reads among various biotypes indicates that the sequencing data effectively captures the complexity of the transcriptome. Conversely, an overrepresentation or underrepresentation of certain biotypes might suggest issues such as biases in library preparation, sequencing depth, or RNA degradation. For example, an unusually high proportion of rRNA reads could indicate contamination with ribosomal RNA, which is typically removed during library preparation. Therefore, interpreting these parameters in the context of the experimental design and known biological expectations is crucial for ensuring the reliability and validity of RNA-Seq data analysis. Additionally, understanding the distribution of different biotypes can provide valuable insights into the regulatory mechanisms and functional dynamics of the transcriptome.
2 - Quality Control for Bulk RNA-Seq data
2.1 - Starting quality control
General assesement in the Project Overview
In the project overview app, you can check the general parameters for your study, such as the sample size or if the important variables are visible in the details tab.
Investigating Alignment statistics
For further statistics about the data integration we need to go into the ‘Sample QC’ app. First, we are presented with the Alignment statistics tab, showing different histograms for quality parameters.
Number of input reads
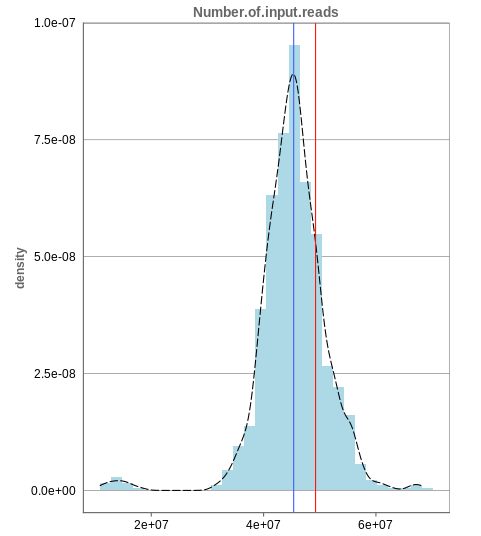
The first histogram is concerned with the number of input reads. While the blue vertical bar shows the median, the red bar shows the position of the current sample selected. A low number of reads can correspond to problematic data, while the definition of “low number” is sometimes subjective and numbers might depend on tissue type or species. Panhunter uses different color codes ranging from white to green to show low to high number of reads in the table below.
While absolute reads may be hard to distinguish by, big jumps should be investigated closely. While most of the data in the picture has 30-60 million reads, there are samples that have half or even less of the input reads. Taking these samples out should yield more homogeneous data which are more meaningful for further analysis.
Unique mapping
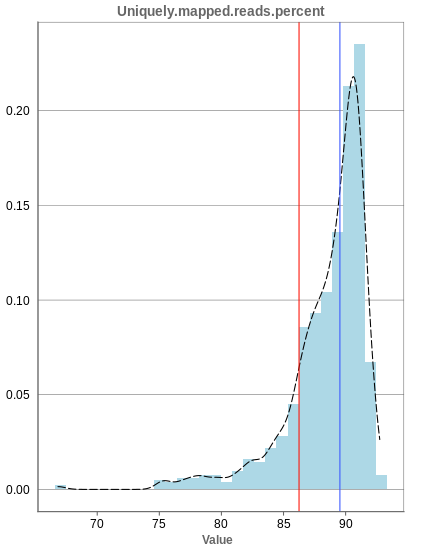
The general plot structure is similar to that of the number of input reads, such that we still want to detect outliers in the histogram. Because non-unique mappings of the input reads are not considered as reads in Panhunter, having a high percentage of uniquely mapped reads is very important.
While the displayed percentage above 85% is good for most samples, there is one with under 70% that should be omitted from the data. This also depends on the type of tissue that was used, such that the given number of 85% is considered meaningful for blood, but when using muscle cells, there might be much more lower numbers.
Mapping to multiple locations
While some reads are uniquely mapped, there is also the case of reads that are not mapped at all and reads that are mapped to multiple locations. As said before, these are not considered in the preprocessing of Panhunter, and only uniquely mapped reads are relevant for the count matrices.
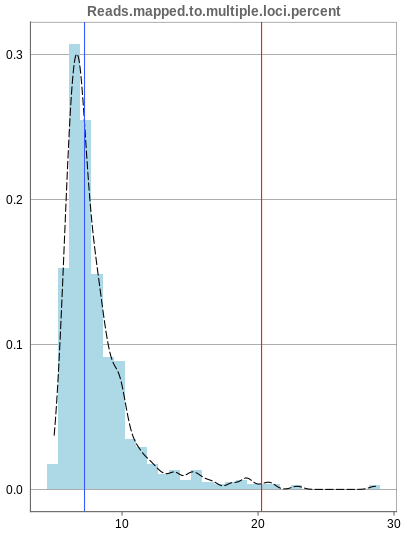
The remaining percentage of reads should largely be composed of multiple mapped reads, i.e. the number of reads that map to no location should be small.
2.2 - Deeper look into SampleQC
There are various other tabs in SampleQC, that are relevant for BulkRNA data.
Read distribution
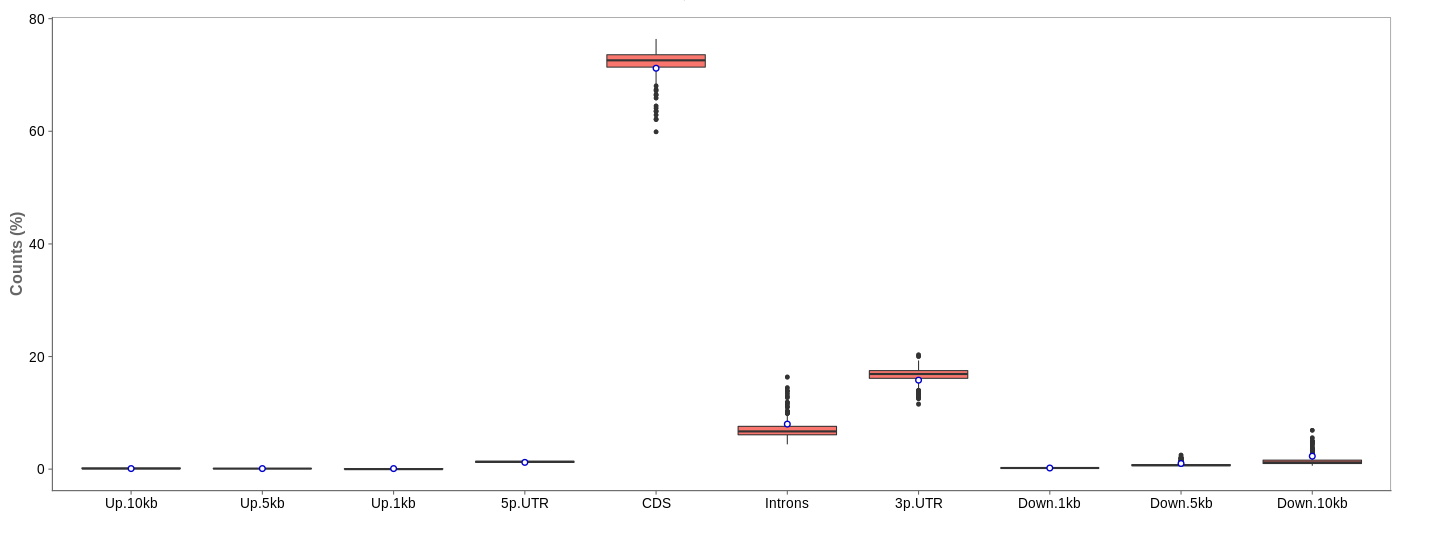
First of all, the box for total percentages should be ticked in the bottom for better comparison.
Ideally, the percentage of counts mapping to the highest should be CDS which stands for coding DNA sequence, relating to the region that codes for protein.
Also important is that up- and downstream are not mapped often.
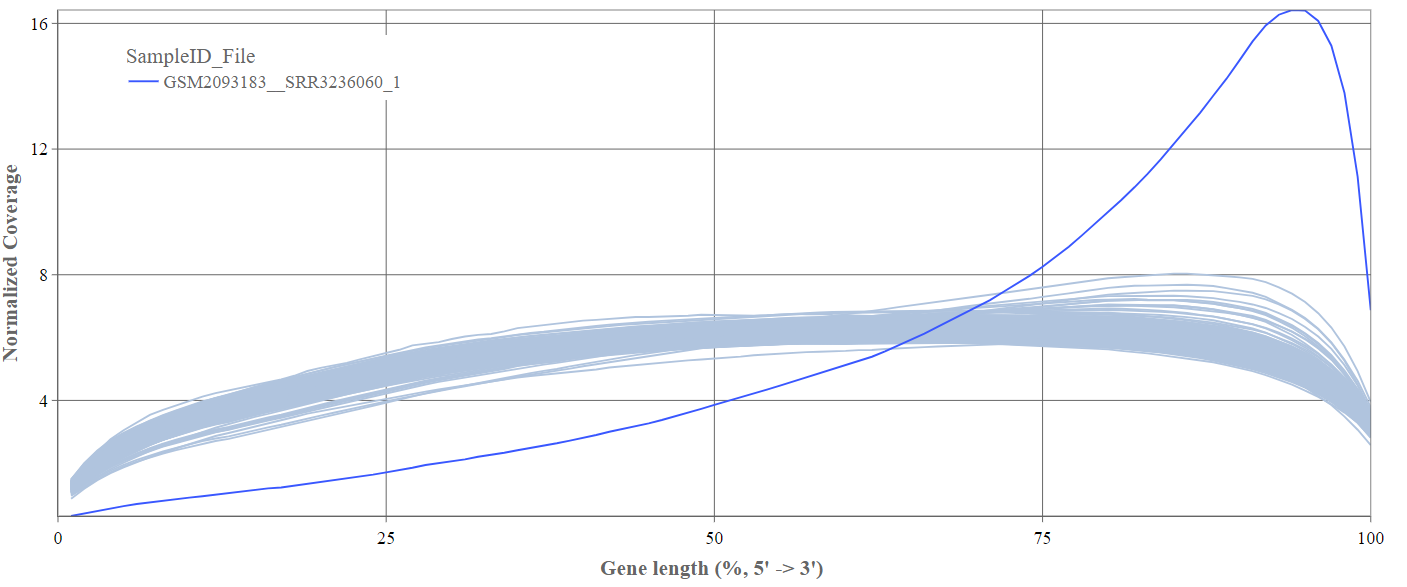
Gene body coverage
We can see the normalized coverage. The samples gene body coverages should be close to one another and should form a homogeneous band and, in BulkRNA-Seq, should be spanning quite ‘uniform’ over the gene length going down at the start and endpoints. Please note that evoenthouth this QC section is called gene body coverage we effectively measure the coverage along the RNA transcripts corresponding to the gene.
The sample colored in blue shows very untypical behaviour and is advised to be excluded.
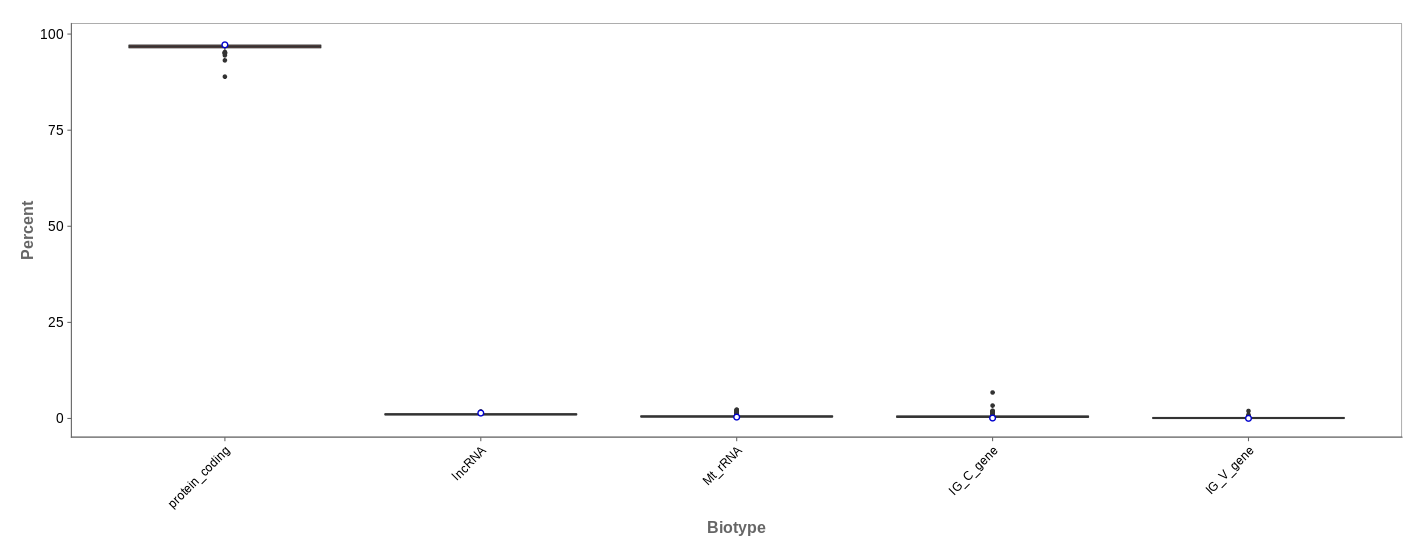
Biotype
The biotype (that is functional types of the genes to which reads are mapped) can be used for quality control. The protein_coding should be the highest and not far away from 100%. We can also see in our example that there we have IgC and IGv genes for tuberculosis data, which seems reasonable because they are relevant for the immune/antibody system of humans.
Mitochondrial
Usually, the proportion of transcripts mapping to mitochondrial genes should be low. If there is phenomenons such as cell death, the amount of mitochondrial transcripts increases and we might infer a lower quality for the data.
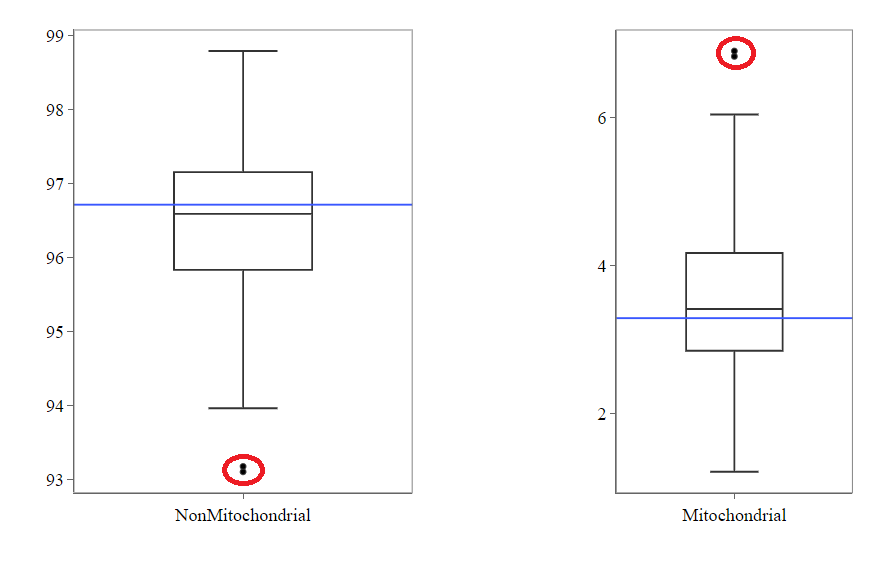
In our example, it may be useful to take out the two circled samples because they are clear outliers from the rest of the data.
Genes
An additional evaluation can be done by looking into the Genes tab, checking which genes are counted the most. The interpretation of this analysis is experiment specific and requires knowledge of the biological system that is investigated.
2.3 - QC clustering
The quality of the data can also be inferred from gene expression profiles represented via dimension reduction methods. Suitable methods in Panhunter are PCA, t-SNE and UMAP. The ‘New Comparisons’ app should be used in the following.
Clustering by sex
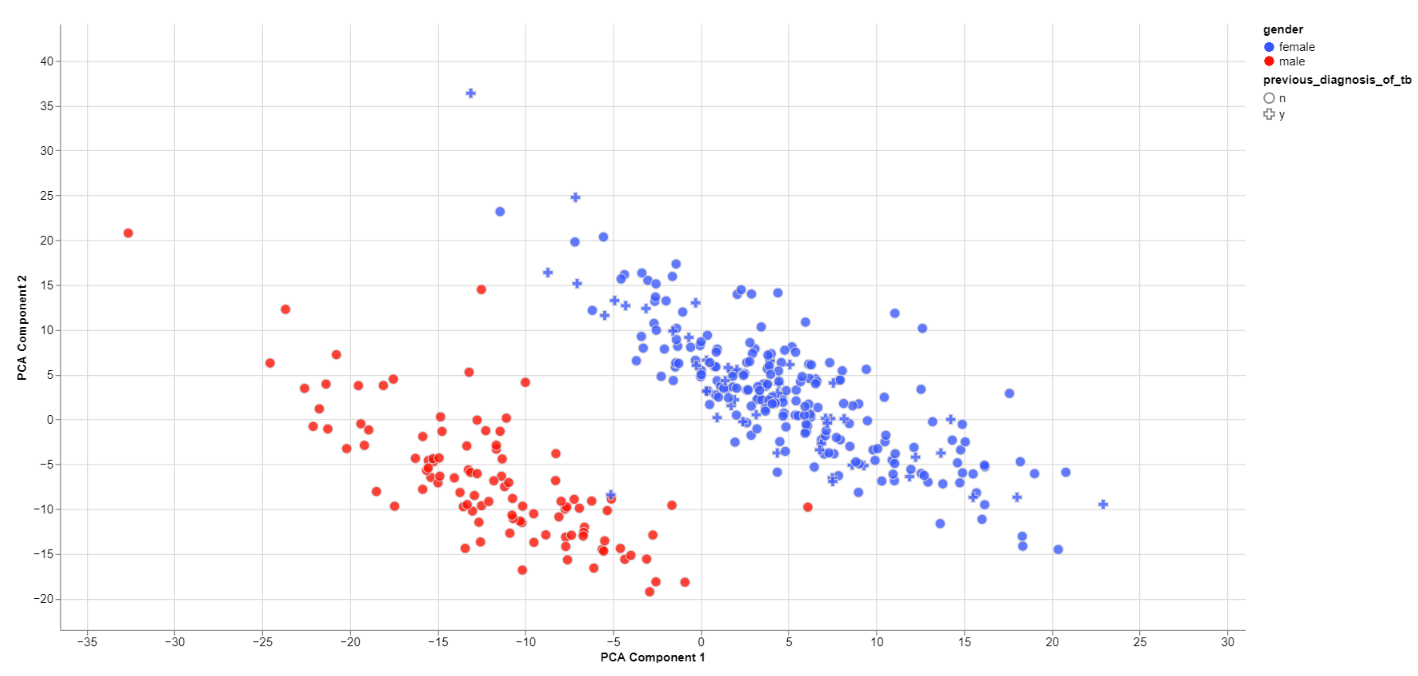
The biological sex should always be one of the greatest disparities between data points because an entire chromosome is different. Thus, there may be reason to exclude samples that are truly different from the rest of their group. However, some studies list gender which should not be confused with the biological sex.
What can also be inferred from the plot is that, in this data set, only females have been diagonsed with tuberculosis before, leading to a dependence between gender and previous_diagnosis_of_tb. These relationships are very important because correlation explained by the previous diagnosis can in some cases be completely explained by the gender/sex association.
PCA features
The categorical and numerical variables that are responsible for the variance in the data can be calculated with the buttons shown below in the picture.
⚠️ Keep in mind that the default is to analyze only the current 2D plot of the dimensional reduction and may vary even by method (PCA,t-SNE, UMAP).
Check outliers
Even when there is no gender/sex variable present, there might be clear clustering. When comparing two groups, one can use the check outliers tab after selecting the groups, to look at up and downregulated genes and infer information from the results.
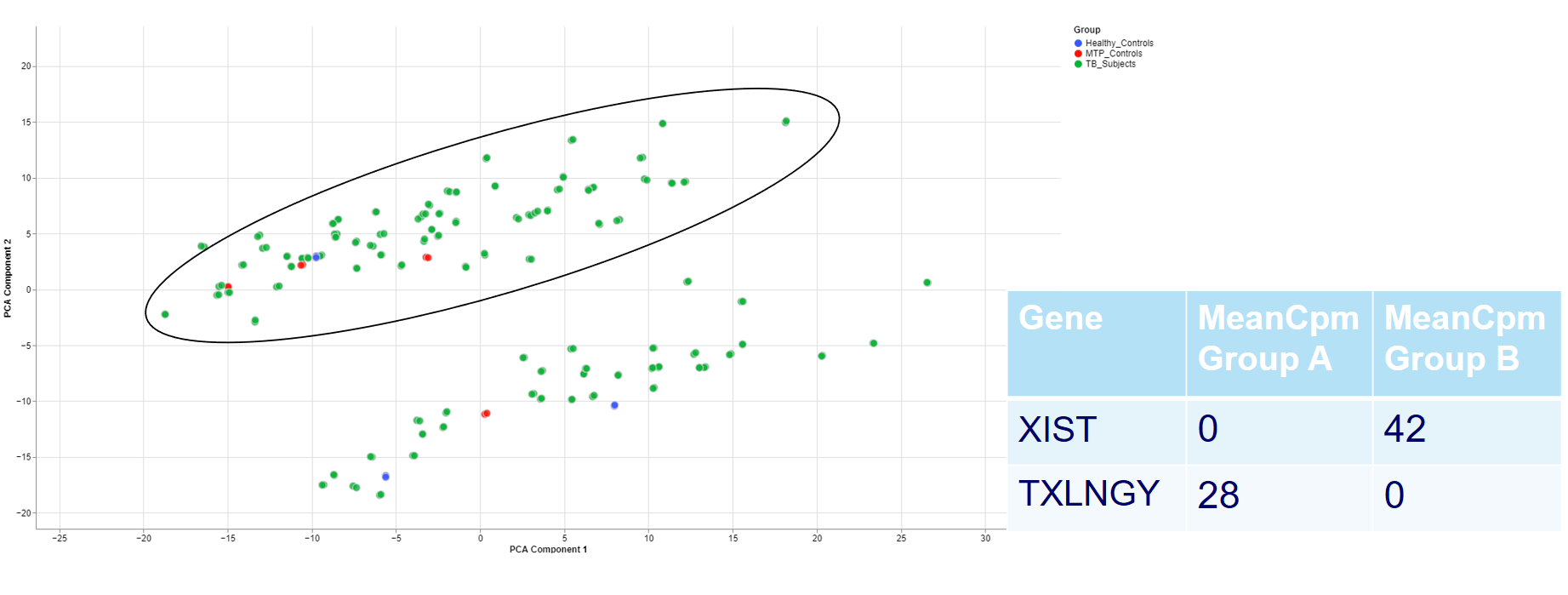
For example, the two groups are clustering completely different in the PCA plot. Even when there is no labeling variable, the comparison shows that certain genes are more present in the groups. Because the presented genes belong to the inactive X chromosome and the Y chromosome, we can infer that there are males and females in the data.
This is important for quality control because of the large influence of the biological sex which might otherwise be neglected.
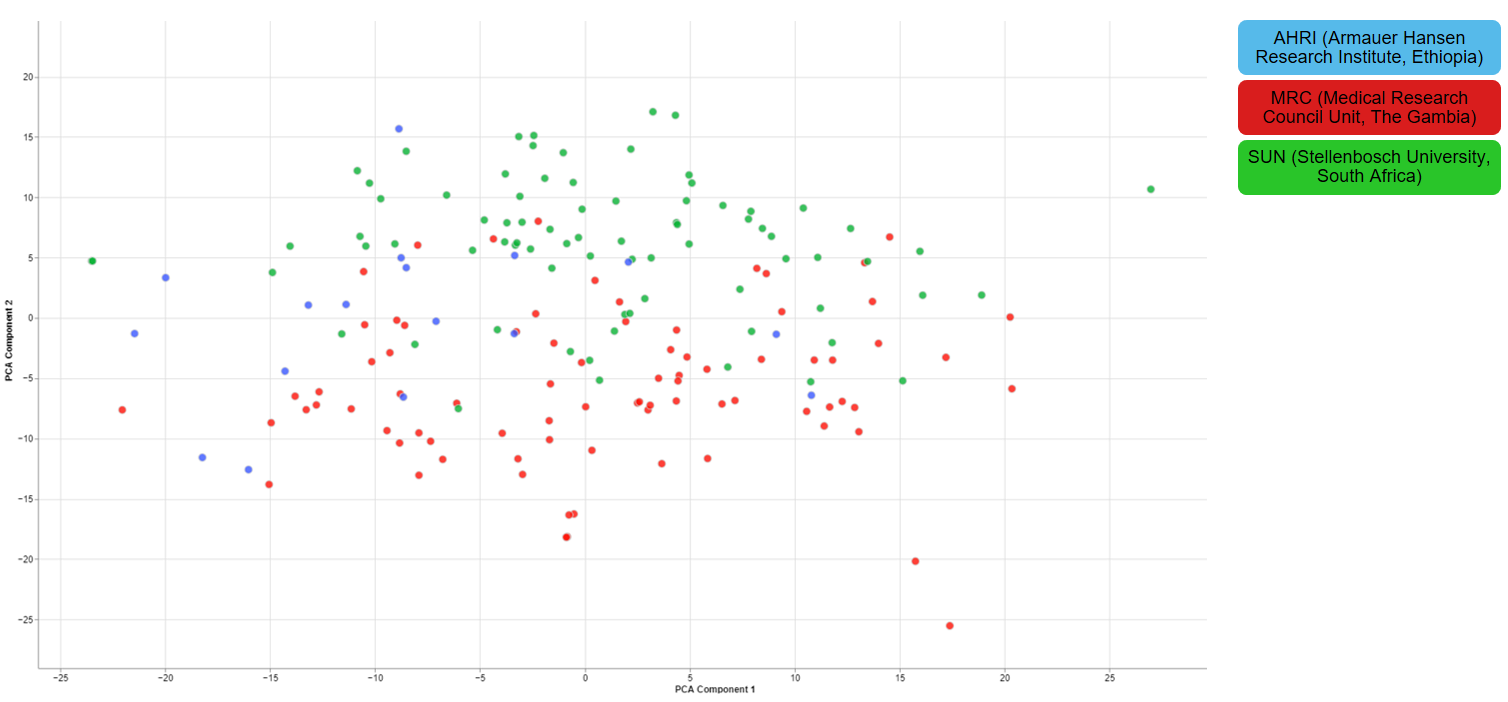
Clustering of other features
For one of the studies, different sites were used to collect blood samples from participants. This inevitable lead to differences that have to be accounted for and are thus important for the quality of the data.
2.4 - Finalizing the Quality Control
There are still various things that can be done to investigate the quality of the data but just a few final thoughts are mentioned down below.
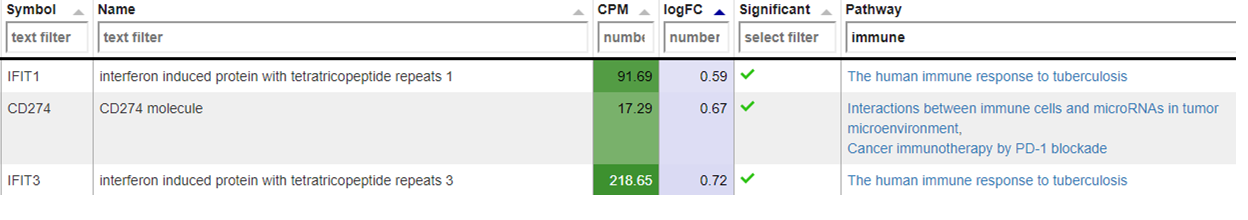
New comparisons
As outlook, one can fit a group comparison in the app ‘New comparisons’, tab ‘New comparison’ with relevant groups, such as TB_subjects against control. If important genes are up/downregulated, then this is another sanity check that can be done.
Reporting on the quality
If there are multiple studies, one could present as follows:
- general setup of the study, things that are noticeable immediately (samples missing)
- general QC stats
- clustering
- variables that are relevant and should be/should not be
- New comparison between important groups, i.e. treatment vs control
- short summary of the study, the QC and important conclusions
- general setup of the next study
- comparison of all studies
- Slide with table: study | sample | reason to take out